
Professor John Rouse FRSE
Scientific Programme Leader (MRC)

Contact
Research
Overview
The chemical reactivity of DNA contributes to the staggering array of DNA lesions that occur in cellular genomes every day. In addition to their potential mutagenicity, these DNA lesions can block important processes such as DNA replication, which can potentially prevent cell proliferation. It is vital that DNA damage is repaired rapidly to prevent mutations, rearrangements or changes in chromosome number from occurring. Cells have evolved sophisticated pathways that repair DNA damage, and stalled or broken DNA replication forks. Defects in these pathways can have serious consequences that range from cell death and embryonic lethality to a range of debilitating disease syndromes.
We are interested in how cells detect, signal and repair DNA damage and how they deal with blocks to DNA replication. In the past years we have discovered a range of factors in mammalian cells that are instrumental for repair of DNA damage and broken replication forks. We are interested in figuring out the modes of action of these proteins and their relevance to disease, and in discovering more new players in DNA repair. Many important chemotherapeutic agents act by inducing DNA damage and/or DNA replication stress and we are interested in finding ways of making these therapies more effective and in preventing resistance. Furthermore we are involved in identifying new anti-cancer drug targets in the DNA repair arena.
Selected research highlights
- Discovery of metazoan SLX4, and demonstrating that it acts as a scaffold for three DNA repair nucleases. SLX4 thus serves as a “molecular toolkit” to process of variety of DNA repair intermediates (2009) (Figure 1)
- Discovery of the human SLX1 nuclease, a Holliday junction resolvase which binds to SLX4. Scientists had been searching for Holliday junction resolvases in human cells for decades but had failed to identify SLX1-SLX4 (2009)
- Discovery of FAN1, a structure-specific nuclease. FAN1 is recruited to sites of DNA damage by the mono-ubiquitinated form of FANCD2, and is required for ICL repair (2011)
- Discovery of the human orthologue of yeast Mms22 (MMS22L) and the associated regulatory subunit TONSL, and the finding that MMS22L and TONSL are important for the repair of broken DNA replication forks (2010)
- Joint discovery in collaboration with the de Winter group that human SLX4 germ line mutations cause Fanconi anaemia as a result of inefficient repair of DNA inter-strand crosslinks (2011)
- Discovery of DVC1, a protein adaptor which recruits to sites of DNA damage the 997-VCP ATPase to facilitate extraction of translesion DNA polymerases from sites of DNA damage to keep mutation rate low in cells (2012)
- Discovery that the SLX1 nuclease acts cooperatively with MUS81-EME1 to mediate almost all processing of Holliday junctions in mitotic mammalian cells (2013)
Current Research Selected projects
A. Characterization of new DNA damage response proteins
We are studying a range of previously uncharacterized open reading frames that are involved in DNA damage responses
B. Mechanisms of action of modes of regulation of DNA repair nucleases
Fanconi anaemia (FA) is a rare inherited chromosome instability syndrome accompanied by developmental and skeletal defects, bone marrow failure and predisposition to cancer. There are fifteen FANC proteins, and the central component of the FA pathway is FANCD2, which is mono–ubiquitylated at Lys561 in S–phase and in response to ICLs. This is catalysed by the eight–subunit FA core complex. The mono-ubiquitylation of FANCD2 is essential for the repair of DNA inter-strand crosslinks (ICLs) but despite much work in this area exactly how mono–ubiquitylation of FANCD2 promotes ICL repair at the molecular level was unknown. In 2010, my lab made a major breakthrough by showing that the UBZ domain of the previously uncharacterized FAN1 protein interacts with the mono-ubiquitylated form of FANCD2, and that FAN1 is recruited to sites of DNA damage in a manner that requires FANCD2 mono–ubiquitylation. FAN1 is a structure–specific nuclease that is specific for 5’ DNA flaps. Intriguingly, like the SLX4 complex, FAN1 appears to process DNA repair intermediates during the HR stage of ICL repair to enable regeneration of an intact replication fork. So binding of FAN1 to mono-ubiquitylated FANCD2 at least partly explains how FANCD2 mono–ubiquitylation regulates DNA repair. However, the available evidence indicates that there must be other ligands of mono-ubiquitylated FANCD2 that regulate ICL repair, and we are searching for these. We are currently using mouse models to decipher the precise mode of action of FAN1 and its regulation by phosphorylation and ubiquitination. We have developed small molecule inhibitors to probe the function of FAN1 and we will use these to test the idea that FAN1 might be a new anti-cancer drug target.
C. The SLX4 complex – a “molecular Swiss army knife” for DNA repair
We refer to the SLX4 complex is a “molecular toolkit” for DNA repair because the three nucleases can cleave a variety of branched DNA species that resemble intermediates of DNA repair (Figure 1).
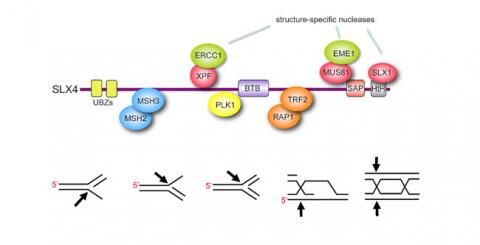
Figure 1 The SLX4 complex: a “molecular Swiss army knife” for DNA repair
Cells depleted of SLX4 are particularly sensitive to agents that cause inter-strand cross-links (ICLs), toxic lesions that block DNA replication forks. The SLX4 complex processes branched intermediates that occur during the HR step of ICL repair required to regenerate an intact replication fork. In collaboration with Johan de Winter (Erasmus Medical Centre, Rotterdam) we discovered that biallelic mutations in SLX4 cause Fanconi anaemia (see below), further underscoring the importance of the SLX4 complex for human health. Recently we identified new functions for the SLX4 complex, apart from repair of inter-strand crosslinks. We showed that some of the SLX4 complex localizes constitutively at chromosome ends (telomeres) (Figure 2).
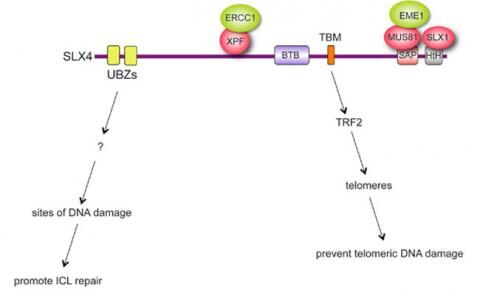
Figure 2 Dual, independent modes of SLX4 localization
This is mediated by a new motif we identified in SLX4 that interacts with the telomere binding protein TRF2. Mutations in this motif release SLX4 complex from telomeres. We also found that the proper localization of SLX4 at telomeres is required to prevent telomeres from becoming too long. Mutations that release SLX4 from telomeres cause telomeres lengthening, and they cause telomere fragility and damage. Holliday junctions are 4-way DNA junctions that intertwine – tangle – chromosomes. They arise normally during DNA repair, and if they are not removed then cells die as they try to segregate their chromosomes in mitosis. It was known that Holliday junction removal requires special nucleases (enzymes that cut DNA) – or “resolvases”. The resolvases in bacteria and phages that remove Holliday junctions are known; they act as homodimers, with each subunit of the homodimer introducing one of the two cuts in Holliday junctions necessary for their resolution. For over 30 years scientists have been searching for the resolvases in eukaryotes that remove Holliday junctions from our DNA in vivo. We recently showed that HJ resolution in eukaryotes requires two separate nucleases - MUS81 and SLX1 - that act together as a HJ resolvase. The two nucleases act cooperatively – the first cut is probably introduced by SLX1 to create the preferred substrate for MUS81, which makes the second cut to finish resolution. Consistent with this idea, both SLX1 and MUS81 nucleases bind close together on the SLX4 scaffold, and this tethering is essential for HJ resolution in cells. So in eukaryotes the two incisions in Holliday junctions necessary to remove them are made by two separate nucleases tethered close together on a scaffold, instead of by the two subunits of a homodimer. We propose this arrangement in eukaryotes increases the ease of regulating resolvase activity. We are currently investigating how the SLX4 complex is regulated by post-translational modification, and new factors that influence the function of the complex.
D. Modulation of DNA repair to improve cancer treatments
We are testing the power of manipulating DNA repair pathways in making chemotherapies more effective in killing cancer cells.
Media availability
I am available for media commentary on my research.
Control of chromosome stability and DNA repair in health and disease
Contact Corporate Communications for media enquiries.
Areas of expertise
- Cancer
Awards
| Award | Year |
|---|---|
| Fellow of the Royal Society of Edinburgh | 2017 |
| National Sciences Prizes awarded since 1990 / The Tenovus Medal 2011 | 2011 |
| National Sciences Prizes awarded since 1990 / The Colworth Medal of the British Biochemical Society | 2009 |
| International Science Prizes awarded since 1990 / EMBO Young Investigator | 2006 |
Stories
- Type
- News
Seven-figure boost for Dundee research
Professor Anton Gartner and Professor Hari Hundal awarded for a joint project with Robert Gordon University.
- Type
- News
John Rouse Wins 2017 Fanconi Anemia Research Fund Discovery Award
John Rouse, together with researchers Detlev Schindler at the University of Wuerzburg, and Minoru Takata at the University of Kyoto, have won the Fanconi Anemia Research Fund 2017 Discovery Award.