
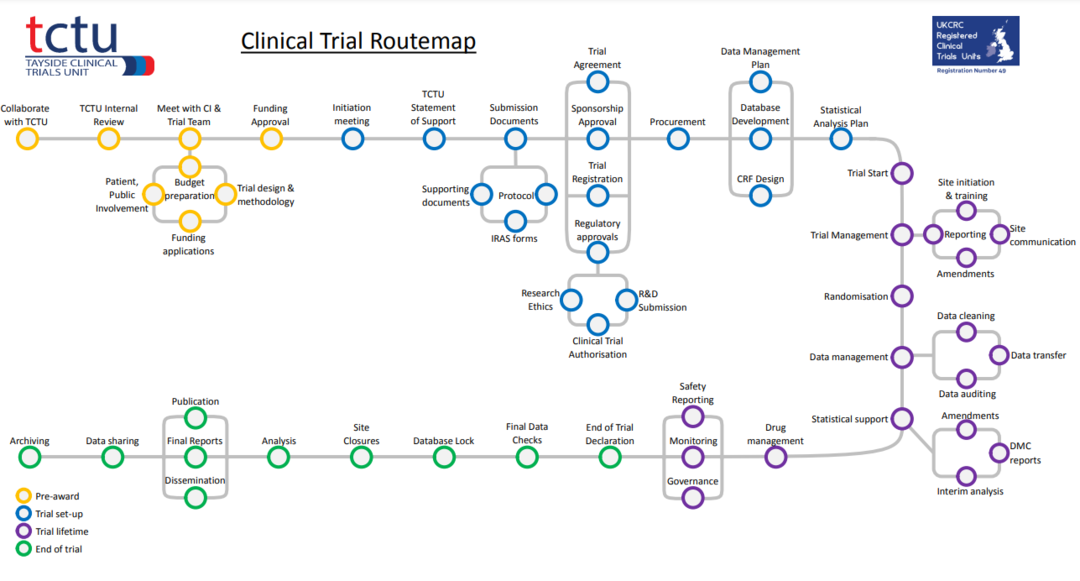
TCTU trial process
Timeline of the clinical trial process followed by TCTU.
Based on the NIHR Clinical Trials Toolkit, we have developed a route map that depicts the lifecycle of a clinical trial.
The route map contains a series of stations, each station on the route map is a key process during the planning, setup, execution and closure of a clinical trial. The stations describe the services and support TCTU can provide at each stage of the trial.
Pre-award
TCTU has a team with a variety of expertise in supporting investigators with Clinical Trials of Investigational Medicinal Products (CTIMPs), device trials, complex intervention trials and qualitative trials.
We recommend you contact us as early as possible when seeking funding for a clinical trial. This enables a collaborative approach to identify all resources to be included at the grant application stage and ensures you have access to the expertise available within TCTU. We can also discuss the design of the trial and offer our input.
Please download and complete (as much as possible) our collaboration form and include any other documents which may be useful to us, such as the draft protocol and draft grant application, and send to TCTU@dundee.ac.uk.
We are happy to help and support you with your funding application and can offer this support without costs to yourself. We will work with you prior to your funding application to determine what support you require from TCTU throughout the lifecycle of your trial and determine costs for this. Whilst we can offer a full package of support from regulatory approval submission, trial management, data management, randomisation, IMP management, and statistical support, you may only need some of these services for your trial.
TCTU will review your proposed trial to see if it requires TCTU input and if TCTU will be willing to provide support. If we feel that you do not require our support or it is not a trial that we would be willing to support, a member of TCTU will let you know with an explanation and offer of advice.
The CI will be invited to meet with the TCTU team. The TCTU team will depend on the type of support required from TCTU and could include a trial manager, data manager, statistician, randomisation programmer, as well as representatives from other stakeholders such as TASC, Health Informatics Centre (HIC), Clinical Research Centre (CRC) or Tayside Pharmaceuticals.
The aims of this meeting are to obtain the key information we need to agree the most appropriate type of TCTU support required to inform budget preparation and to review the design and methodology of the trial. Example discussion areas:
- Confirmation of funder and submission deadlines
- TCTU role and responsibilities
- TCTU resource required
- Involvement of other stakeholders: Sponsor, TASC involvement (NHS, Clinical Research Centre, Tissue Bank, HIC etc.)
- Project and trial timelines
- Trial design and methodology
- Patient pathway
- Outcome measures
- Sample size calculations
- Feasibility
TASC SOP 56 v5 The Preparation and Peer Review of Sample Size Calculation for Clinical Research
3.1 Budget preparation
TCTU will provide support and advice for the identification and preparation of costs for your clinical trial. We can provide costs for TCTU resources and liaise with other departments for their costs, for example NHS costs. Prior to submission for funding a Project Registration Form (PRF) must be submitted to Research Finance Services for final costs and approval of the School Dean.
3.2 Trial design and methodology
Along with budget preparation, TCTU can help with reviewing the design, including participant pathway, and methodology of your trial, and provide statistical help with producing power calculations.
3.3 Submission of funding application
Please ensure TCTU are aware of the submission deadline and leave enough time for PRF sign off.
3.4 Patient & Public Involvement
It is important to involve patients, carers and the public in prioritising, designing and delivering health research studies. PPI is important to ensure that research studies are planned to answer important questions for patients and to ensure research designs are appropriate and accessible to the patient population. Many funders look for PPI activities and budgets to be built into grant applications. TCTU can advise you on PPI activities that can add value to your study, including study design, documentation, recruitment strategies, acceptability, accessibility and relevance, as well as sense making of results and dissemination of results in a meaningful way.
For PPI support from the TASC PPI Manager, please contact public-involvement@dundee.ac.uk
TASC SOP 66 Planning Patient and Public Involvement (PPI) In Clinical Research
Please let TCTU know as soon as possible if funding has been approved or not. If funding has not been approved we can work with you for any resubmissions you wish to make. If funding has been approved, we will arrange to meet with you to discuss timelines and TCTU support.
Trial setup
Following a successful funding application, TCTU will organise a meeting with the CI’s research team together with any other relevant stakeholders.
TCTU will provide a Statement of Support which details the services to be provided by TCTU to the Chief Investigator. This could include submission to regulatory authorities, trial management, data management, statistical support, randomisation system and or drug management system. A Statement of Support will be put in place before TCTU commences work on the trial.
Where it has been agreed that TCTU will support you with your regulatory submissions, we work with you to draft and/or review all required documents.
Protocol
TCTU can work with you to draft the protocol using information from the funding application or to review any protocol that you may have written. This would include various members of TCTU, for example Trial Manager, Data Manager, Statistician, and others outside TCTU, such as Clinical Trial Pharmacists as required.
TASC SOP 14 Writing a protocol to Good Clinical Practice for Clinical Trials of Investigational Medicinal Products
TASC SOP 47 The use of Version Control of Study Documents used in Health and Social Care Research Studies
TASC Policy 11 Co enrolment
Supporting documents
TCTU can draft and/or review these for you. These include:
- Participant Information Sheet, Informed Consent Form, Participant Invite Letters, GP Letters, and Questionnaires
- Organisation Information Document (OID)
- Schedule of Events Cost Attribution Template (SoECAT)
IRAS forms (Integrated Research Application System)
TCTU can complete this form for the CI to review and authorise.
Approvals
TASC SOP 18 Trial Set Up in Clinical Trials of Investigational Medicinal Products
- Doc Ref 018 TMF Index v9
- Doc Ref 019 ISF Index v6
- Doc Ref 057 Delegation Log v9
- Doc Ref 071 Participant Randomisation Log v5
- Doc Ref 076 Screening Log v5
- Doc Ref 118 PSF Index v2
TASC Policy 10 Clinical Research Projects Involving Human Tissue
Sponsorship Approvals
TCTU can help with this submission and liaise with the Sponsor over any changes they may wish to be made.
TASC SOP 28 Sponsorship of Clinical Trials and TASC SOP29 Application for Sponsorship of Health and Social Care Research Studies (excluding Drug and Device Studies)
TASC SOP 29 Application for Sponsorship of Health and Social Care Research Studies (excluding Drug (e.g., CTIMP) and Device Studies)
Research Ethics
TCTU can assist with the submission to the Research Ethics Committee (REC) through the Integrated Research Application System (IRAS). Following submission, the Chief Investigator will be invited to attend a REC meeting. TCTU can also attend this meeting if required.
Following a REC meeting, the Chief Investigator will be notified of the decision, with one of the following outcomes:
- Favourable opinion
- Favourable opinion with conditions
- Provisional opinion
- Unfavourable opinion
Where a favourable opinion with conditions, provisional opinion or unfavourable opinion is given we can work to address the REC concerns and resubmit any documentation they require.
R&D Submission
TCTU can assist with the submission to NHS R&D offices.
Clinical Trial Authorisation
TCTU can assist with the submission to Clinical Trial Authorisation (CTA) from the Medicine and Healthcare Products Regulatory Agency (MHRA).
Trial Agreements
TCTU can liaise with the TASC Legal Team to confirm the appropriate agreements between the Sponsor, the Chief Investigator and external stakeholders are in place.
TASC Policy 07 Contracts
TASC Policy 12 Selection and Oversight of Vendors for Clinical Research
Trial Registration
For CTIMPs, TCTU can complete the mandatory registration in the European Clinical Trials Database (EudraCT). For other trials we can register in another approved database, for example clinicaltrials.gov or International Standard Randomised Controlled Trial Number Registry (ISRCTN).
TASC SOP 61 Registering and Reporting Research in a Publicly Accessible Database
TASC Policy 06 Study Registration & Publication
TASC Policy 07 Contracts
TASC Policy 12 Selection and Oversight of Vendors for Clinical Research
TASC SOP 34 Implementation and Maintenance of a Quality Assurance System in Clinical Research Laboratories
TASC SOP 35 Management of a Chain of Custody for Samples in Clinical Research
TASC SOP 36 Preparation of an Analytical Plan for Laboratories Associated with Clinical Research
TCTU can liaise with the University of Dundee procurement to ensure that any procurement conforms to the relevant legislation.
TASC Policy 12 Selection and Oversight of Vendors for Clinical Research
The Data Management Team can provide different levels of data management support depending on your requirements, which will be discussed and costed for at the pre-award stage. The level of support we offer ranges from an oversight package where we will advise you on best practise all the way through to a full Data Management package which would include:
- Creation of a DMP
- CRF design
- DMS design, build and validation
- DMS maintenance
- User training for data entry staff
- User management
- Data quality checks
- Data auditing
- MedDRA coding of adverse events
- SAE reconciliation with the sponsor Pharmacovigilance department
- Database lock
- Liaise with the statistician/health economist to establish their data requirements and provision of data to them
- Liaise with site staff to ensure timely data entry and resolution of data queries
- Provide reports to trial team to assist with trial management and monitoring
- Provide data for Data Monitoring Committee
- Data sharing
- Data archiving
We can also provide a secure online data transfer system to facilitate the transfer of files, including laboratory results, force plate data, actometer data to TCTU for incorporation into the final trial datasets.
Data Management Plan
The Data Management Team can assist you in writing a Data Management Plan (DMP) or generate one for you. The DMP describes and provides details of the data management processes that will be employed throughout the lifecycle of the trial. Some funders require a DMP at the application stage so it is important to set aside some time to think about what data you will collect, the volume of data you will collect, where the data will be generated, for example participant, laboratory, MRI, medical notes, and how you will keep the data secure.
TASC SOP 48 Data Management in Clinical Research Studies using Excel
TASC SOP 53 Data Management in Clinical Research
Database Development
The Data Management Team can provide you with a web-based, GCP–compliant Data Management Software (DMS) to record the data collected during the trial. Key requirements of GCP-compliance in terms of computerised systems in clinical trials include system validation, full audit trail capabilities, individual user accounts, data security, and data backups. The design of the DMS will be carried out alongside the design of the CRF and will incorporate point of entry data checks. A key part of the validation activities is releasing the system for user acceptance testing (UAT). This allows you to examine the system prior to it ‘going live’ for data entry and ensure it meets your requirements. The UAT may be carried out by various roles including the Trial Manager, researcher or research nurse.
The majority of the work carried out by the Data Management Team is focussed later on during the course of the trial after data collection has started and is centred around data quality and provision of data to relevant parties after database lock. For more information, see Database Management, Database Lock, Data Sharing and Data Archiving for more information.
TASC Policy 09 Clinical Research Computer System Validation
TASC SOP 48 Data Management in Clinical Research Studies using Excel
TASC SOP 53 Data Management in Clinical Research
Case Report Form Design
The Case Report Form (CRF) or electronic Case Report Form (eCRF) is an essential document which will be used to record the data collected for each trial participant during the trial. The CRF standardises data collection across sites and is defined in the protocol. The Data Management Team can assist you with CRF design, liaising with you and other stakeholders (statistician and Trial Management Team) to create a CRF for your trial.
TASC SOP 19 Preparing and maintaining case report forms (CRF) for use in Clinical Research
TCTU statisticians can work with you before the trial begins to initiate a Statistical Analysis Plan (SAP) for the trial. This will describe the study design, analysis population, sample size calculation, the proposed statistical methods that will be used and the data collection and management procedures.
TCTU statisticians will also have input during the trial if any amendments are made that will affect the final analysis.
TASC SOP 05 v12 Statistical Analysis Plans for Clinical Research
TASC SOP 57 v5 Implementation of Statistical Analysis in Clinical Research
Trial lifetime
Once all trial documentation has been finalised and approved by regulatory bodies the trial can begin! Trial sites can be set up, become active and start recruiting participants.
TASC SOP 18 Trial Set Up in Clinical Trials of Investigational Medicinal Products
TCTU can provide different levels of trial management support depending on your requirements. What level of support you need/want will be discussed and costed for at the pre-award stage. This will include the level of staff and time to be spent on the trial. Our Trial Management Team consists of Trial Managers, Trial Co-ordinators and Trial assistants.
Full trial management support
Depending on the size and complexity of your trial a Trial Manager or Trial Co-ordinator will be appointed to project manage your trial during trial setup, recruitment, follow-up and closure. Trial management will include:
- Finalising regulatory approvals
- Ensuring required documents are in place prior to trial start, for example Trial Master File, Working Practice Guidelines, and Delegation Logs
- Liaising with Data Management Team on design of Case Report Forms and data management system
- Setting up randomisation system
- Equipment and trial medication procurement and co-ordination of supplies to sites
- Monitoring recruitment and retention rates and promoting activities to help with both
- Liaising with Data Management Team and Statisticians for data cleaning and statistical reports
- Site initiation and training
- Liaising with TASC governance, monitoring and pharmacovigilance sections
- Liaising with external departments, for example laboratories, imaging, trial medication suppliers
- Setting up and managing trial oversight committees
- Submitting reports, for example REC, funder
- Preparing for audits and inspections
- Site closures
- Final reports
TASC SOP 17 Preparing and Participating in a Regulatory Inspection
TASC SOP 45 Establishing and Maintaining a Trial Master File, Investigator Site File and Pharmacy Site File for use in Clinical Research
- Doc Ref 018 TMF Index v9
- Doc Ref 019 ISF Index v6
- Doc Ref 107 NCTIMP Study Master File Index v3.0
- Doc Ref 118 PSF Index v2
TASC SOP 54 Training Records
TASC SOP 59 Reporting Breaches in Clinical Research
TASC Policy 05 GCP Training for Personnel Involved in Clinical Research
Site Initiation & Training
Trial management will include:
- Identifying sites and working with them to obtain the appropriate regulatory approvals
- Site set-up including trial documentation and equipment supplies
- Training: protocol, trial assessments, Case Report Form, randomisation system, recruitment tracker
- Day-to-day communication with sites for support and problem solving
- Ensure all trial staff are trained for their role
- Coordinating trial drug delivery and storage
TASC SOP 07 Receiving Informed Consent from Potential Participants in Clinical Research
TASC SOP 11 Identifying, Recording and Reporting Adverse Events for Clinical Research
- Doc Ref 058a Pregnancy Notification v5
- Doc Ref 058b Pregnancy Follow-up v5
- Doc Ref 072 SAE Form v18
- Doc Ref 086 AE Log v6
TASC SOP 12 Establishing Identity of Participants in Clinical Research
TASC SOP 23 Completion of Delegation Log
TASC SOP 45 Establishing and Maintaining a Trial Master File, Investigator Site File and Pharmacy Site File for use in Clinical Research
- Doc Ref 018 TMF Index v9
- Doc Ref 019 ISF Index v6
- Doc Ref 107 NCTIMP Study Master File Index v3.0
- Doc Ref 118 PSF Index v2
TASC SOP 59 Reporting Breaches in Clinical Research
TASC Policy 05 GCP Training for Personnel Involved in Clinical Research
Site Communication
The Trial Management Team will maintain contact with sites throughout the duration of the trial and provide advice and guidance. The Trial Management Team will submit the necessary regulatory amendments and approvals and work to set up and initiate new sites.
Amendments
Where necessary changes to the trial are identified by yourself TCTU will advise if the changes affect the regulatory approvals and on any amendment submissions required for these changes. The trial team will draft the required changes to any approved documents and submit to Sponsor for review. Once approved and classified by Sponsor the Trial Management Team will submit to the regulatory authorities. The Trial Management Team will review other documents and systems to identify any changes required and liaise with the Data Management Team if the changes will have an impact on their work. Once the amendment has been fully approved the Trial Management Team will work with the sites to ensure all appropriate documentation is shared and complete any training on the changes which may be required.
TASC SOP 63 Amendments to Healthcare Research Projects
Reporting
The Trial Management Team will work with you to draft any regulatory reports, for example REC Annual Progress Report, and will liaise with the Sponsor Pharmacovigilance team for submission of annual MHRA Development Safety Update Reports if appropriate. The Trial Management Team will also support you in submitting any reports required by the funder. They will also work with the Data Management Team and Statisticians if any reports are required for oversight committees such as a Data Monitoring Committee.
TASC SOP 15 Preparing and Submitting Reports for Clinical Research
TASC SOP 65 Preparing and Submitting Development Safety Update Reports (DSUR) for Clinical Trials of Investigational Medicinal Products (CTIMP)
TCTU have worked with the Health Informatics Centre, University of Dundee to develop the Tayside Randomisation System (TRuST). TRuST is a GCP-compliant randomisation and drug management interactive web response system (IWRS) suitable for CTIMP, non-CTIMP and complex intervention trials.
TRuST offers a range of functions to support a clinical trial, including: randomisation, IMP allocation, IMP accountability, IMP request forms, site IMP supply monitoring, remote monitoring and emergency unblinding.
We can work with you to develop the specification for your randomisation system. This may be a simple system randomly allocating participants to a group. However, TRuST can also build in minimisation, stratification, multiple randomisations for the same participant and cluster randomisations. Randomisation may be blinded or unblinded. Where blinded, an unblinding facility is available.
TRuST can also incorporate a drug management system, please see Drug Management station.
TASC SOP 40 Randomisation, Blinding and Code Breaking in Clinical Research
During a trial, the Data Management Team can perform data cleaning/discrepancy management, data coding (for example MedDRA coding of adverse events), data auditing (quality control), Serious Adverse Event reconciliation with the Sponsor Pharmacoviligance department, database lock, data transfer and archiving.
TASC SOP 17 Preparing and Participating in a Regulatory Inspection
TASC SOP 48 Data Management in Clinical Research Studies using Excel
TASC SOP 53 Data Management in Clinical Research
TASC SOP 55 Creating Reports for the Independent Data Monitoring Committee
TASC Policy 09 Clinical Research Computer System Validation
Data cleaning
The overall aim is to generate a high quality dataset for analysis. This is achieved in part by checking the data for logical consistency, missing, incorrect or implausible data. The Data Management Team can carry out data cleaning for you, generating queries which will go back to site for resolution.
Data Transfer
For practical reasons, not all trial data is entered into the DMS; examples might include immunohistochemistry data from biopsies, blood results or MRI data. This type of data may require to be transferred in a ‘batch’ or for all trial participants at the end of the trial. This data will need to be incorporated into the trial datasets for analysis.
The Data Management Team can provide and manage a secure, web-based GCP-compliant system to facilitate this transfer and will liaise with you and the relevant data provider(s) to gather specific requirements. They can also carry out reconciliation of the incoming data with what is expected.
Data Auditing
Data auditing ensures the data recorded in the DMS is accurate. This is important not only in terms of analysis but also in terms of participant safety. The Data Management Team will work with you to plan and carry out auditing activities during the course of your trial and prior to database lock, or alternatively provide advice as to how to carry out audits.
TASC SOP 48 Data Management in Clinical Research Studies using Excel
TASC SOP 53 Data Management in Clinical Research
TCTU statisticians can provide support throughout the trial, including producing reports for data monitoring committees, performing interim analysis, producing a Statistical Analysis Plan (SAP) and providing input into any amendments to the study.
TASC SOP 05 Statistical Analysis Plans for Clinical Research
TASC SOP 55 Creating Reports for the Independent Data Monitoring Committee
TASC SOP 57 Implementation of Statistical Analysis in Clinical Research
Tayside Randomisation System (TRuST) has the functionality for drug management. For drug management, clinical trial pharmacies will have access to TRuST, and drug accountability will be logged from receipt in pharmacy, dispensing to participants and for return and disposal of trial drug. Once a participant is randomised to receive an investigational medicinal product (IMP) TRuST can generate prescription forms, drug accountability logs, destruction notices and similar. TRuST can be used for complex dosing regimens, calculating dose requirements for individual participants, for example depending on weight and dose titration. The Trial Management Team will work with the drug supplier and, using TRuST, ensure timely delivery of stock to sites.
In discussion with yourself, TCTU will develop the specification for TRuST.
TASC SOP 37 Accountability, Returns and Destruction of Investigational Medicinal Products in CTIMPs
TASC SOP 38 v9 Manufacturing, Assembly, Packaging and Labelling of Investigational Medicinal Products in Clinical Trial Investigational Medical Products
TASC SOP 39 Supply, Transport and Storage of Investigational Medicinal Products in Clinical Trial Investigational Medical Products
TASC SOP 43 Handling Product Recalls of Clinical Trial Investigational Medicinal Products or Other Trial Related Drugs
TCTU can work with you to ensure the correct safety reporting procedures are carried out. We can draft/review the safety reporting section of the protocol to ensure safety reporting is relevant to the trial and conforms to regulatory requirements. We can then work with you to ensure adverse events are recorded, reported, and followed up correctly, not only for regulatory requirements but also to ensure the correct data is recorded for your trial analysis.
TASC SOP 11 Identifying, Recording and Reporting Adverse Events for Clinical Research
- Doc Ref 058a Pregnancy Notification v5
- Doc Ref 058b Pregnancy Follow-up v5
- Doc Ref 072 SAE Form v18
- Doc Ref 086 AE Log v6
Monitoring
Where the Sponsor has deemed that monitoring of the trial is required, for example CTIMPs and high risk non-CTIMPs, TCTU will work with the monitoring team to draft the initial Monitoring Plan. When the trial is underway we can liaise with the monitoring team regarding their site visits and any actions to be followed up.
TASC SOP 03 Monitoring Clinical Trials of Investigational Medicinal Products (CTIMPs)
Governance
TCTU work closely with TASC governance to ensure the trial is performing to current regulations and will liaise with governance regarding the reporting of breaches to protocol, GCP and GDPR.
TASC SOP 59 Reporting Breaches in Clinical Research
End of trial
TCTU can draft and submit your End of Trial Declaration to Sponsor, REC and MHRA as appropriate, within the required timelines.
TASC SOP 16 Closure of Health and Social Care Research Studies
The Data Management Team will perform final checks on the data prior to the final database audit. This gives the CI and statistician assurances on the quality and integrity of the data for analysis.
Database lock is a controlled procedure that prevents further changes to the trial data. This occurs once all the trial data has been entered, all cleaning procedures completed, all queries resolved, safety data reconciled (for example, pharmacovigilance reports), final database audit carried out. After database lock the trial data will be provided to the trial statisticians for analysis. The Data Management Team will carry out these procedures for you if they are providing the DMS, or alternatively can provide advice if you are using another system.
TASC SOP 32 Locking Clinical Study Databases
TCTU Trial Management Team will ensure correct procedures are followed to ensure each trial site is closed.
TASC SOP 16 Closure of Health and Social Care Research Studies
Publication
TCTU can work with you to draft a dissemination plan including dissemination to trial participants.
The TCTU statistician and Trial Management Team will provide input into any papers to be submitted for publication, presentations and similar.
TCTU must be acknowledged for their support either by having named authors or in the acknowledgement section of all publications.
Final Reports
TCTU statisticians and trial management can work with you to complete final reports, such as for funder, REC, or publicly accessible database.
Dissemination
TCTU can assist you in sharing trial results with the target patient population, in an accessible and understandable format.
TASC SOP 61 Registering and Reporting Research in a Publicly Accessible Database
TASC SOP 66 Planning Patient and Public Involvement (PPI) in Clinical Research
TASC Policy 06 Study Registration & Publication
TCTU can establish a data sharing access committee and facilitate sharing of data to other parties post publication.
TCTU can work with you to ensure all trial documentation, paper and electronic, is archived according to Sponsor SOP for the required length of time.
TASC SOP013 v12 Archiving Clinical Studies